3. Dimensionality reduction
Petr V. Nazarov, LIH
2024-10-14
You should have loaded the following data:
Mice- mouse phenome dataset (790 mice)miR&mRNA- microRNA and mRNA profiles of A375 cell line after stimulation with IFNg (adapted from [Nazarov et al, Nucleic Acid Research, 2013])
If not, please load:
## load the script from Internet
source("http://r.modas.lu/readAMD.r")
## Mouse 'phenome' data
Mice = read.table("http://edu.modas.lu/data/txt/mice.txt",
sep="\t",
header=TRUE,
stringsAsFactors = TRUE)
## miRNA data
miR = readAMD("http://edu.modas.lu/data/txt/mirna_ifng.amd.txt",stringsAsFactors = TRUE)
## mRNA data
mRNA = readAMD("http://edu.modas.lu/data/txt/mrna_ifng.amd.txt",
stringsAsFactors = TRUE,
index.column="GeneSymbol",
sum.func="mean")3.1. Overview of dimensionality reduction methods
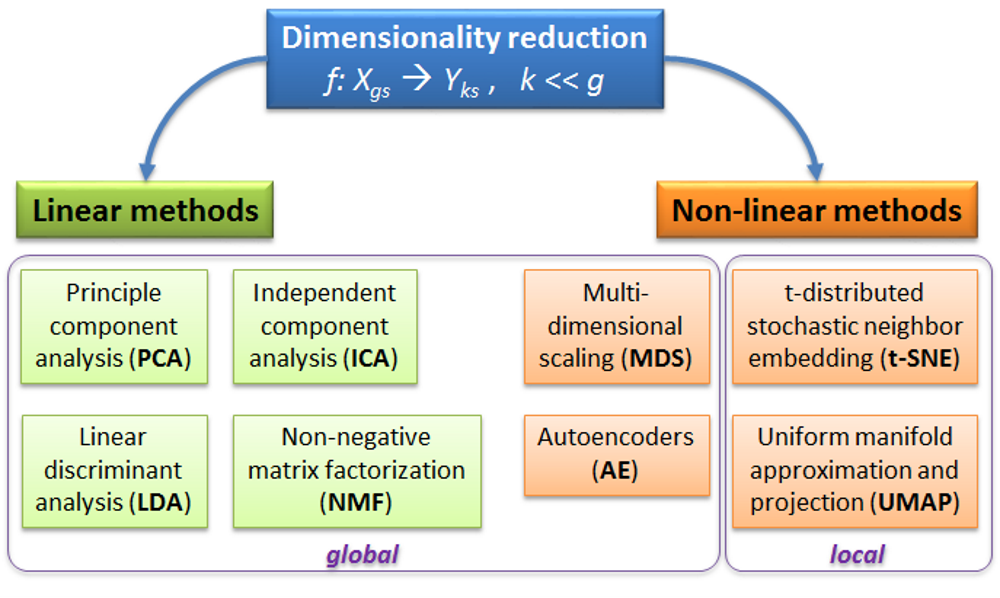
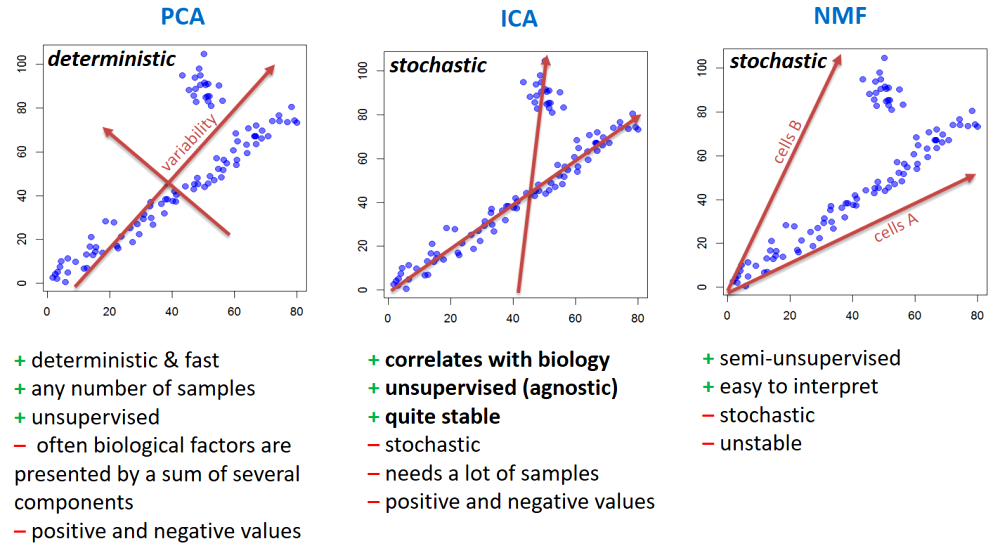
PCA - rotation of the coordinate system in multidimensional space in the way to capture main variability in the data.
ICA - matrix factorization method that identifies statistically independent signals and their weights.
NMF - matrix factorization method that presents data as a matrix product of two non-negative matrices.
LDA - identify new coordinate system, maximizing difference between objects belonging to predefined groups.
MDS - method that tries to preserve distances between objects in the low-dimension space.
AE - artificial neural network with a “bottle-neck”.
t-SNE - an iterative approach, similar to MDS, but consideting only close objects. Thus, similar objects must be close in the new (reduced) space, while distant objects are not influecing the results.
UMAP - modern method, similar to t-SNE, but more stable and with preservation of some information about distant groups (preserving topology of the data).
{kind=link}
Let’s consider some nice resources online:
Principal Component Analysis Explained Visually by Victor Powell
Understanding UMAP by Andy Coenen and Adam Pearce
Dimensionality Reduction for Data Visualization: PCA vs TSNE vs UMAP vs LDA by Sivakar Sivarajah
3.2. PCA - Principle Component Analysis
3.2.1 PCA of samples in the space of features (genes/proteins)
Simplest way to do PCA in R is to use default prcomp()
function. It takes a matrix with samples in rows and features in
columns.
- If you assume that absolute values of features is not important
(only relative variability) - use
scale=TRUE.
PC = prcomp(t(mRNA$X),scale=TRUE)
str(PC)## List of 5
## $ sdev : num [1:17] 83.4 55.3 48.1 42 36 ...
## $ rotation: num [1:20141, 1:17] 0.001125 0.000754 0.006407 0.008881 0.009878 ...
## ..- attr(*, "dimnames")=List of 2
## .. ..$ : chr [1:20141] "" "A1BG" "A1CF" "A2BP1" ...
## .. ..$ : chr [1:17] "PC1" "PC2" "PC3" "PC4" ...
## $ center : Named num [1:20141] 6.6 6.25 5.1 4.64 5.92 ...
## ..- attr(*, "names")= chr [1:20141] "" "A1BG" "A1CF" "A2BP1" ...
## $ scale : Named num [1:20141] 0.0233 0.1059 0.1061 0.0862 0.2095 ...
## ..- attr(*, "names")= chr [1:20141] "" "A1BG" "A1CF" "A2BP1" ...
## $ x : num [1:17, 1:17] -84.9 -104.8 -69.3 -81.6 -70 ...
## ..- attr(*, "dimnames")=List of 2
## .. ..$ : chr [1:17] "T00.1" "T00.2" "T03.1" "T03.2" ...
## .. ..$ : chr [1:17] "PC1" "PC2" "PC3" "PC4" ...
## - attr(*, "class")= chr "prcomp"plot(PC$x[,"PC1"],PC$x[,"PC2"],col=as.integer(mRNA$meta$time),pch=19,cex=2,
main="PCA (scaled gene expression)")
Alternatively you could use my warp-up function
plotPCA(). Let’s try it with and without gene scaling
source("http://r.modas.lu/plotPCA.r")
par(mfcol=c(1,2))
plotPCA(mRNA$X,col=as.integer(mRNA$meta$time),cex.names=1,cex=2,
main="PCA (gene scaling)")
plotPCA(mRNA$X,col=as.integer(mRNA$meta$time),cex.names=1,cex=2, scale=FALSE,
main="PCA (no scaling)")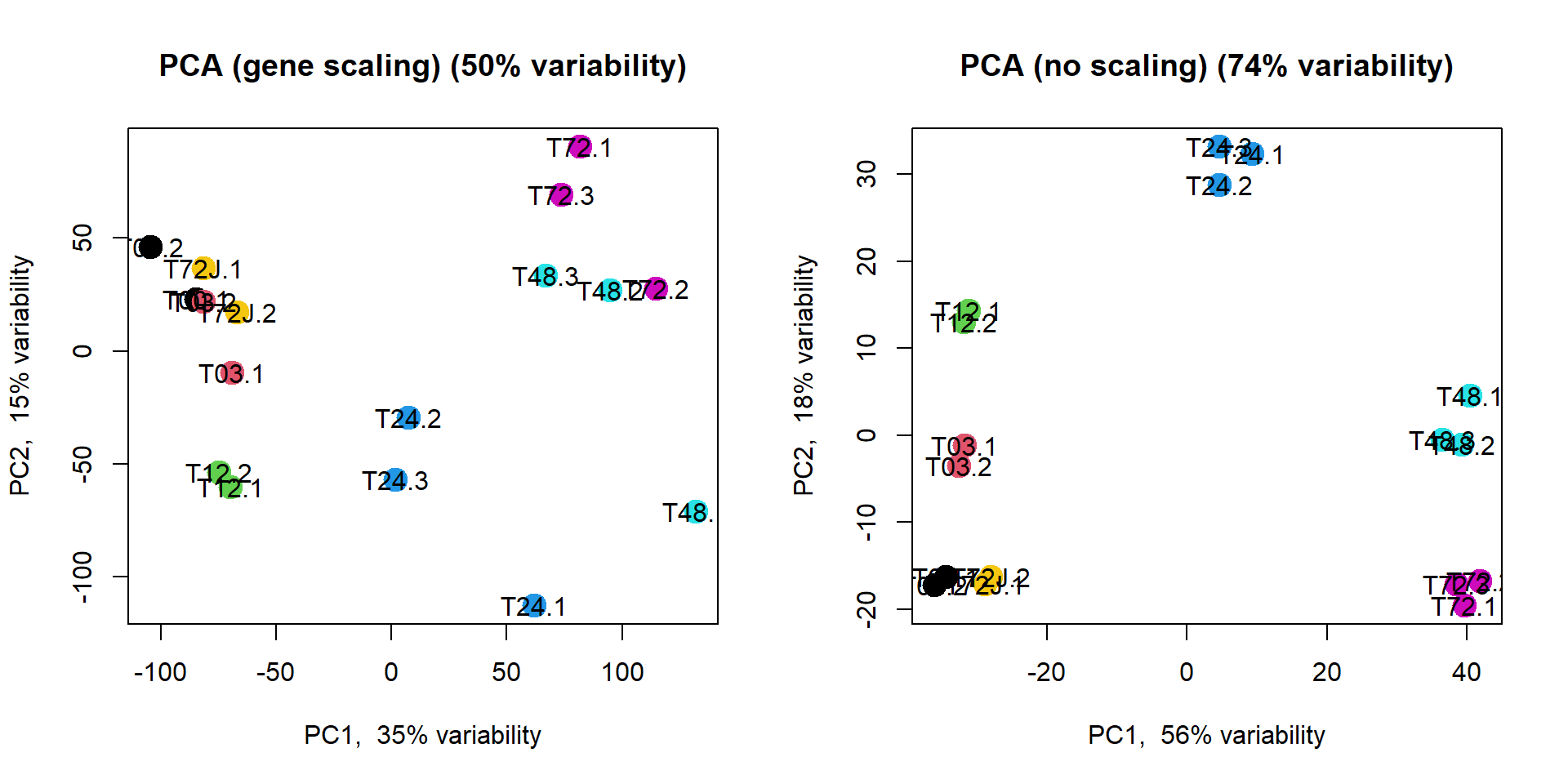
Variability captured by each component can be calculated and visualized as a scree plot manualy:
## calculate percentage of variability
PC$pvar = (PC$sdev^2)/sum(PC$sdev^2)
names(PC$pvar) = colnames(PC$x)
## draw scree plot
barplot(PC$pvar[1:10],ylim = c(0,0.4), col="skyblue",main="Explained Variability",cex.main=1, xlab="Components",ylab="Variability explained")
## add text
text(1.2 * (1:10)-0.5,PC$pvar[1:10],sprintf("%.3f",PC$pvar[1:10]),adj=c(0.5,-1))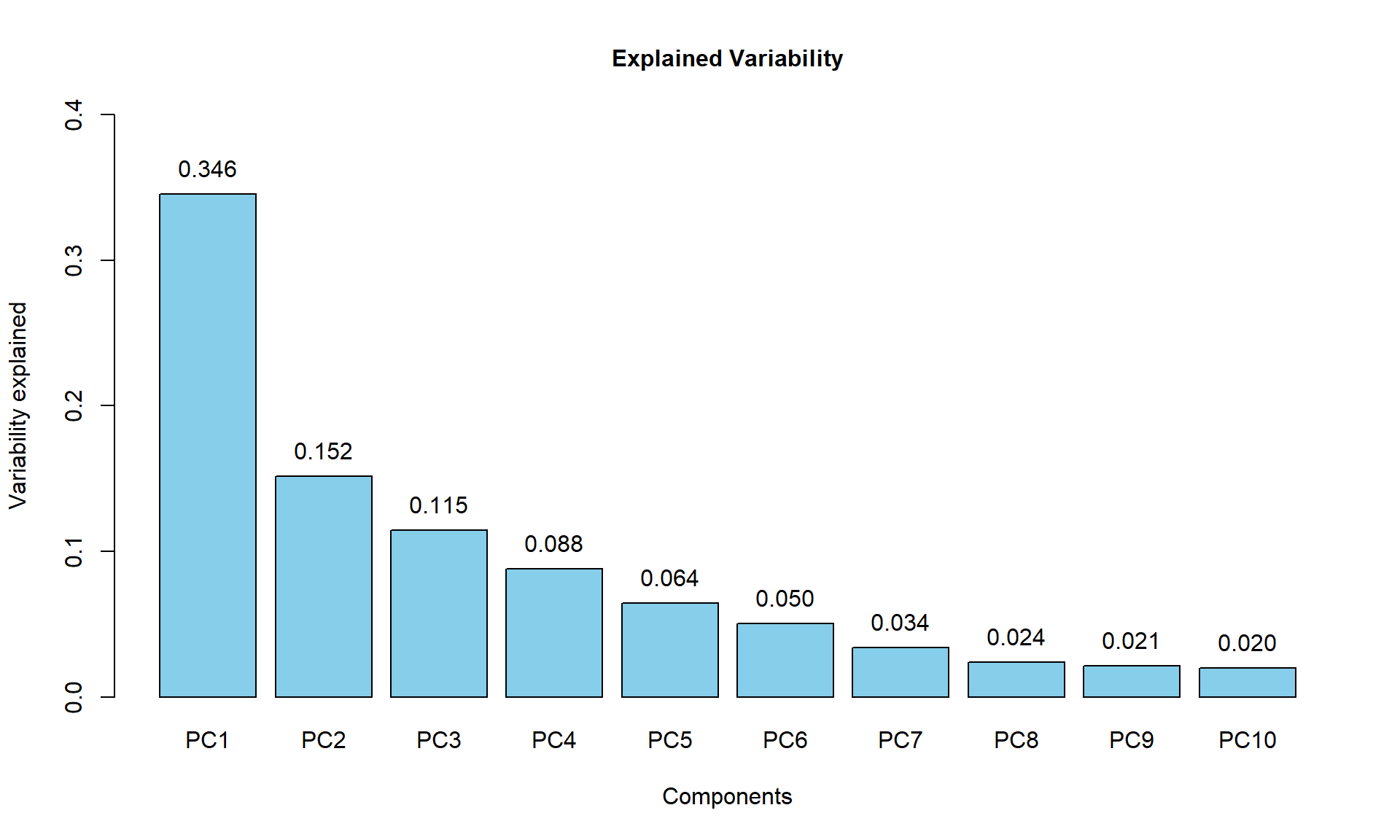
or using a package::function factoextra::fviz_eig() (try
it, if you wish).
factoextra::fviz_eig(PC, addlabels = TRUE, main="Explained Variability (fviz_eig)")
Another powerful package: PCAtools
## please install: PCAtools and ggalt
#install.packages("BiocManager")
#BiocManager::install("PCAtools")
#install.packages("ggalt")
library(PCAtools)
p = pca(mRNA$X, removeVar = 0, metadata = mRNA$meta)
biplot(p, colby="time", title="Principal Component Analysis", legendPosition = "bottom", showLoadings = TRUE,encircle=TRUE)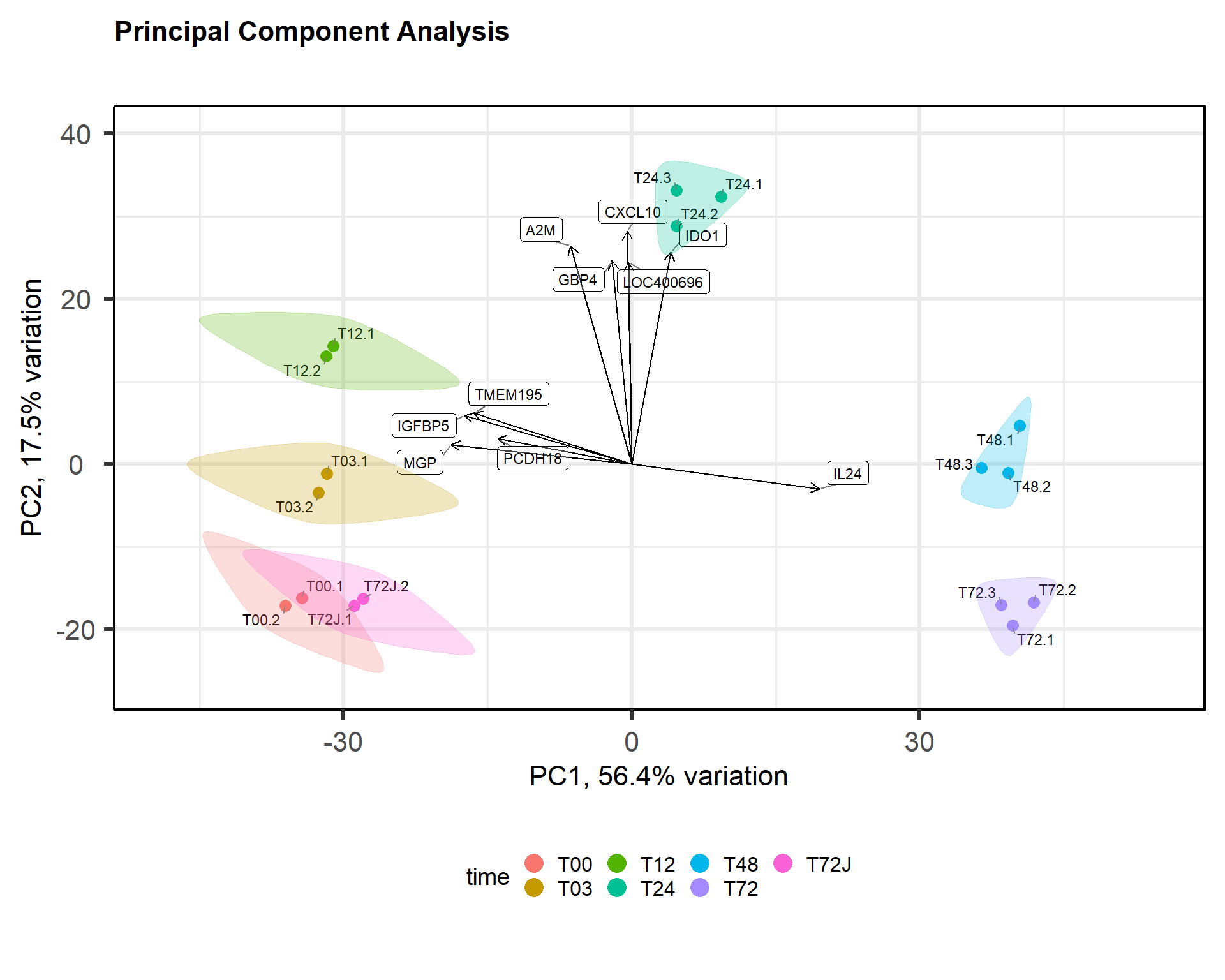
screeplot(p, axisLabSize = 8, titleLabSize = 22)
3.2.2 PCA of genes in the space of samples
We can also use PCA to see similarity between features. Let’s also highlight marker genes.
size = 0.5 + apply(mRNA$X,1,mean)/6
## Log intensities
plotPCA( t(mRNA$X),
col="#00AA0022",
cex=size,
main = "PCA for genes (sample space)")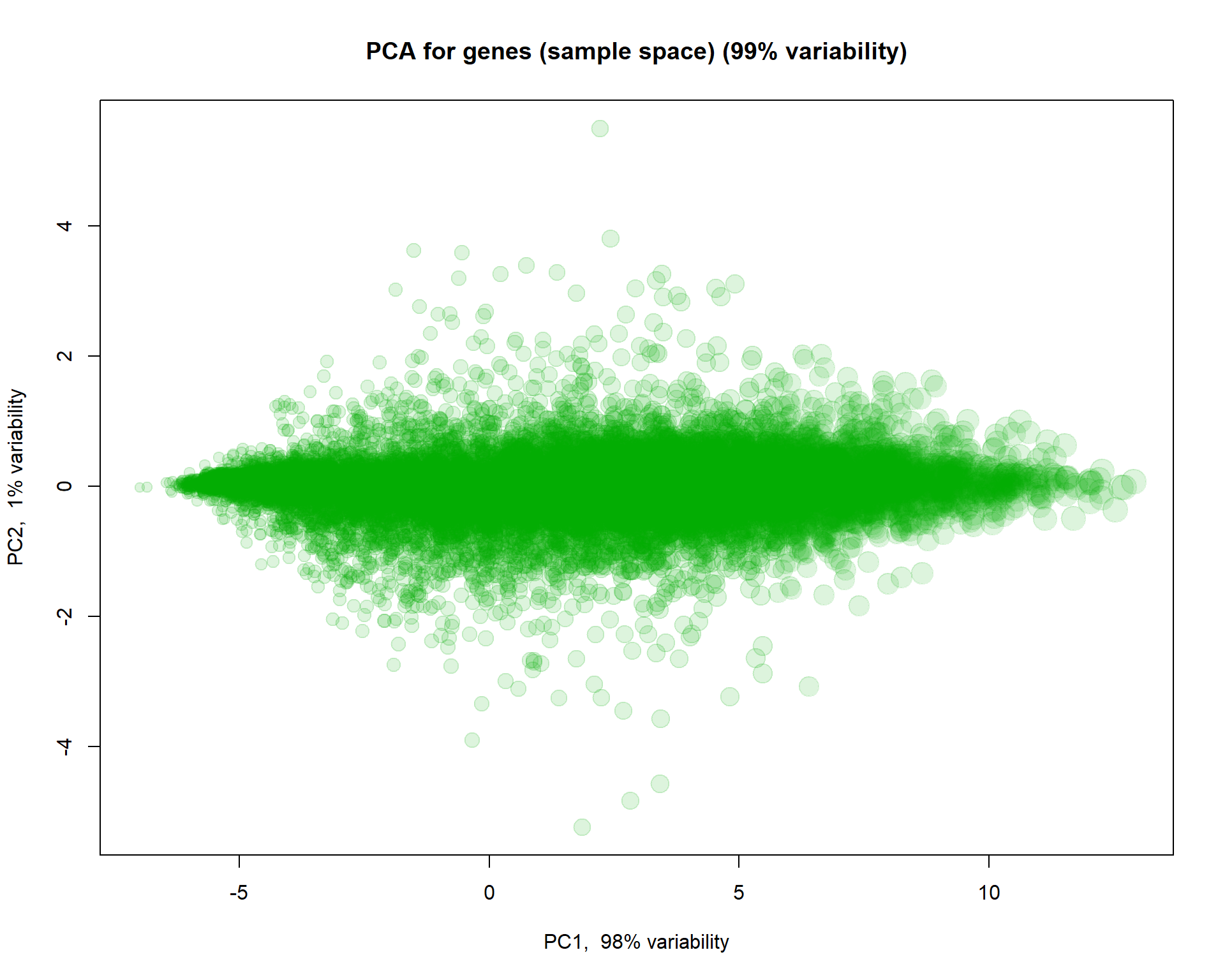
## you can check behaviour of with treatment "VEGFA","IRF1","SOX5This is not too informative, as 98% of variability is related to overall gene expression. Let’s normalize genes (each gene will have mean = 0, st.dev = 0 over samples).
## Let's scale expression
Z = mRNA$X
Z[,] = t(scale(t(Z)))
PC2 = prcomp(Z,scale=TRUE)
plotPCA(t(Z),pc = PC2,col="#00AA0011", cex=size, main = "Standardized expression (per gene)")
## select 3 genes and show them
genes = c(which.min(PC2$x[,1]),which.max(PC2$x[,1]),which.min(abs(PC2$x[,1]-2) * abs(PC2$x[,2]-1)))
text(PC2$x[genes,1],PC2$x[genes,2],names(genes),col=1,pch=19,font=2)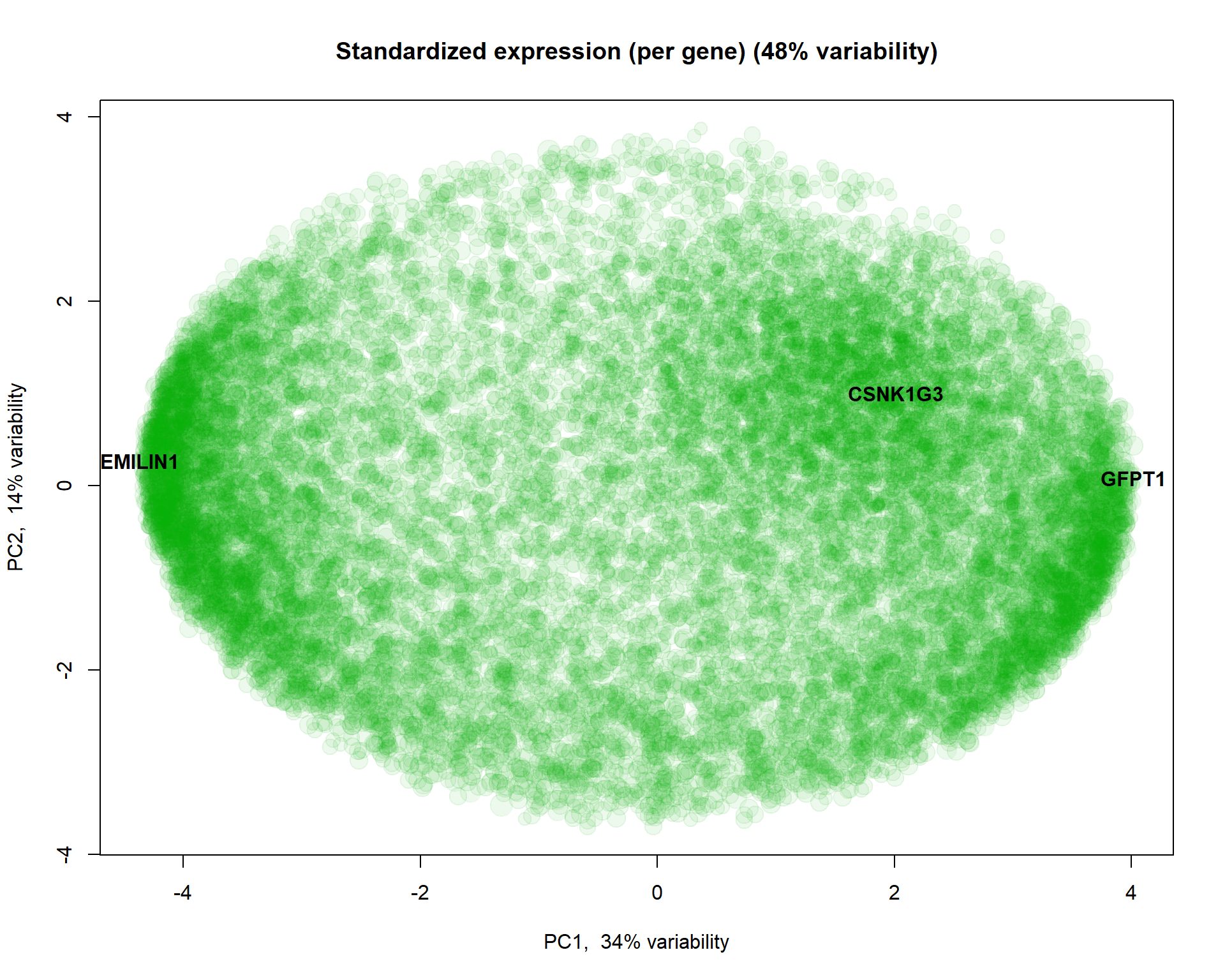
I selected here 3 genes for a detailed investigation:
## behaviour with treatment
par(mfcol=c(1,3))
boxplot(mRNA$X[genes[1],]~mRNA$meta$time, main = names(genes)[1], col = 1:nlevels(mRNA$meta$time))
boxplot(mRNA$X[genes[2],]~mRNA$meta$time, main = names(genes)[2], col = 1:nlevels(mRNA$meta$time))
boxplot(mRNA$X[genes[3],]~mRNA$meta$time, main = names(genes)[3], col = 1:nlevels(mRNA$meta$time))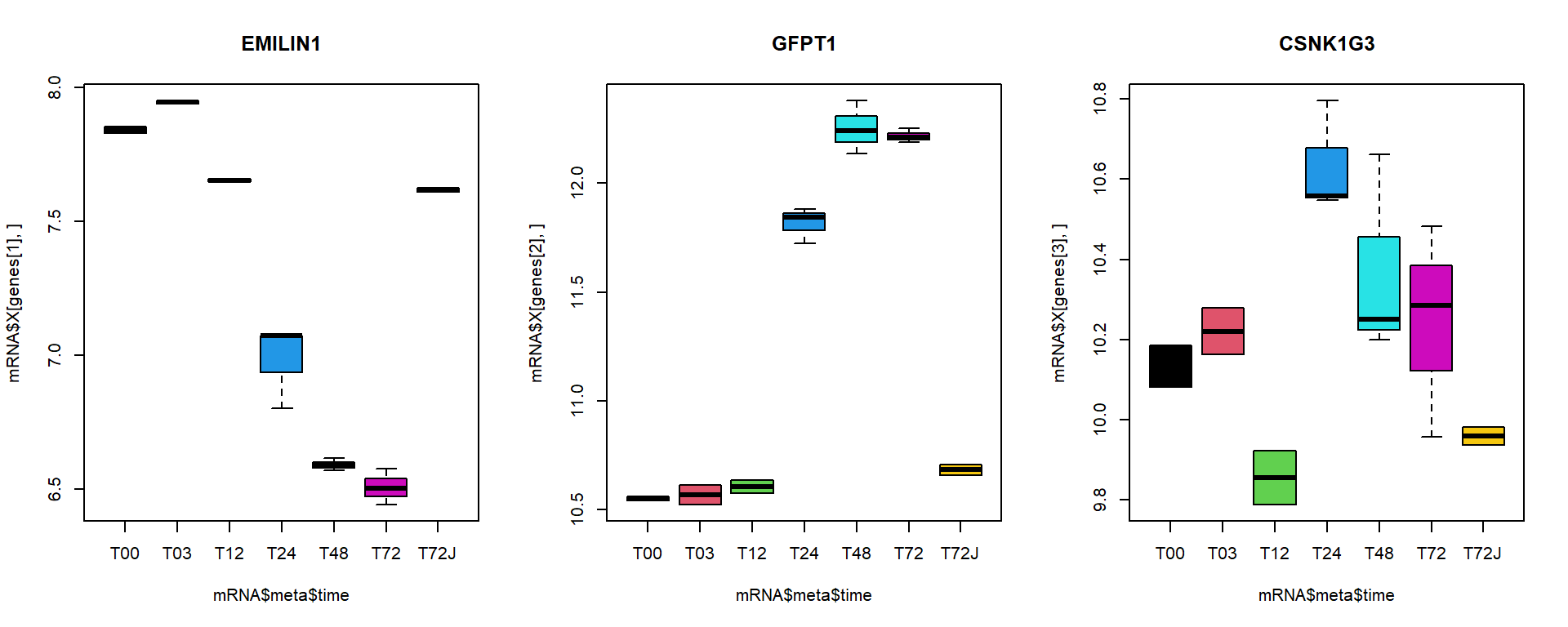
3.3. MDS
Classical MDS can be performed using cmdscale()
function
## calculate euclidean distances
d = dist(t(mRNA$X))
## fit distances in 2D space to distances in multidimensional space
MDS = cmdscale(d,k=2)
str(MDS)## num [1:17, 1:2] -34.4 -36.1 -31.8 -32.6 -31.1 ...
## - attr(*, "dimnames")=List of 2
## ..$ : chr [1:17] "T00.1" "T00.2" "T03.1" "T03.2" ...
## ..$ : NULLplot(MDS[,1],MDS[,2],col=as.integer(mRNA$meta$time),pch=19, main="MDS plot",cex =2)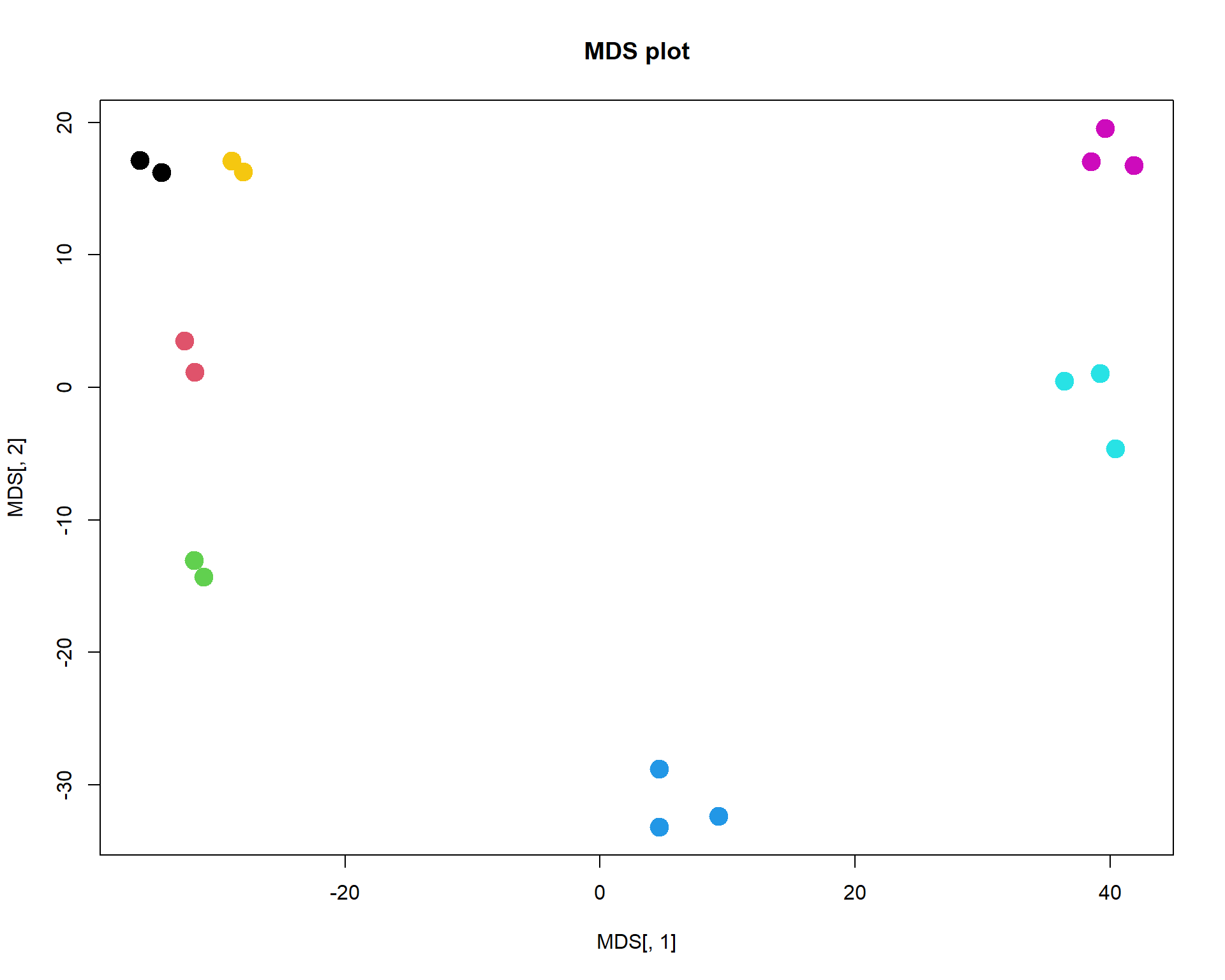
As you can see, it is very close to PCA results. But it requires much more computational power, when working with large number of objects. It may outperform PCA in a special case, when variance captured by 1st, 2nd and 3rd components is similar. Or when you need a special type of distance.
3.4. ICA
ICA, similar to PCA, but tries to extract statistically independent signals. This signals are correlated to biological processes more often than for PCA. But to see ICA power we need a lot of data. Here results will be similar to MDS.
source("http://r.modas.lu/LibICA.r")## [1] "----------------------------------------------------------------"
## [1] "libML: Machine learning library"
## [1] "Needed:"
## [1] "randomForest" "e1071"
## [1] "Absent:"
## character(0)
## [1] "----------------------------------------------------------------"
## [1] "consICA: Checking required packages and installing missing ones"
## [1] "Needed:"
## [1] "fastICA" "sm" "vioplot" "pheatmap" "gplots"
## [6] "foreach" "survival" "topGO" "org.Hs.eg.db" "org.Mm.eg.db"
## [11] "doSNOW"
## [1] "Absent:"
## character(0)IC = runICA(mRNA$X,ntry=100,ncomp=3,ncores=2)## *** Starting parallel calculation on 2 core(s)...
## *** System: windows
## *** 3 components, 100 runs, 20141 features, 17 samples.
## *** Start time: 2024-10-15 18:05:07.077469
## *** Done!
## *** End time: 2024-10-15 18:05:16.784222
## Time difference of 9.684211 secs
## Calculate ||X-SxM|| and r2 between component weights
## Correlate rows of S between tries
## Build consensus ICA
## Analyse stabilitypar(mfcol=c(1,3))
plot(IC$M[1,],IC$M[2,],pch=19,col=as.integer(mRNA$meta$time),cex=2,main="IC1-IC2")
plot(IC$M[1,],IC$M[3,],pch=19,col=as.integer(mRNA$meta$time),cex=2,main="IC1-IC3")
plot(IC$M[2,],IC$M[3,],pch=19,col=as.integer(mRNA$meta$time),cex=2,main="IC2-IC3")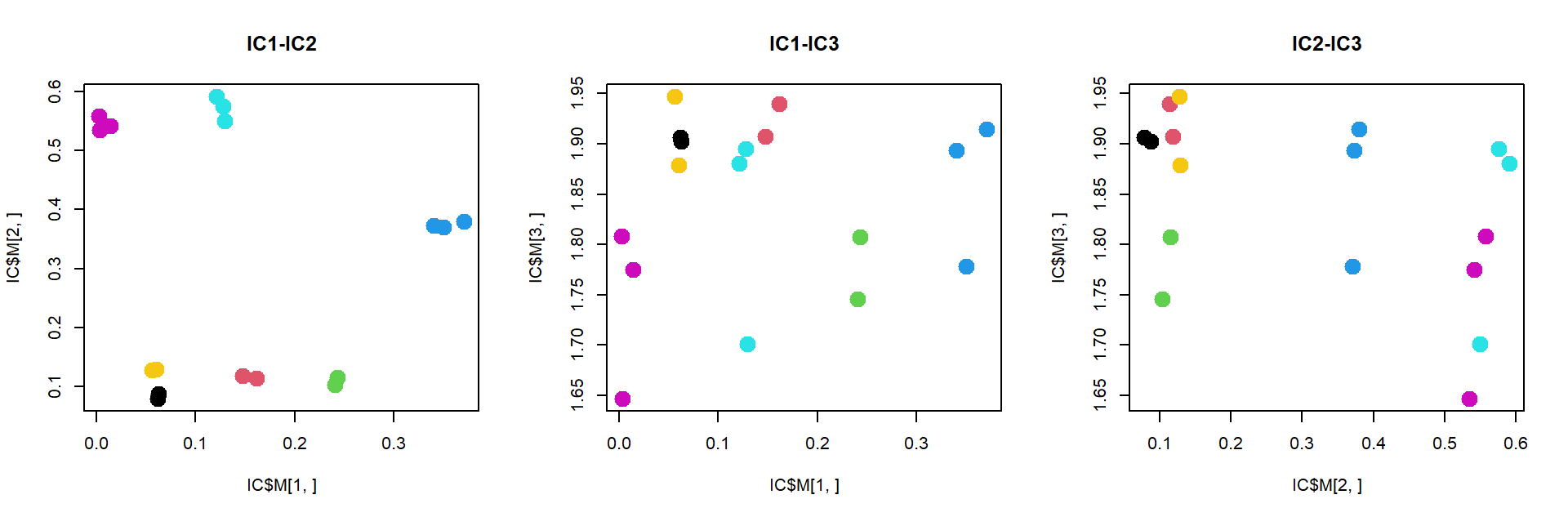
3.5. t-SNE
Unlike PCA, t-SNE shows not differences, but rather similarities.
Similar objects should be located together. The method does not care
about distant objects. The distance is weighted by Student (t)
distribution (similar to Gaussian). Let’s use Rtsne
package. Please note, that when your dataset is large, it is recommended
to do PCA first before sending your data to t-SNE. In Rtsne
they use so called “Barnes-Hut implementation” - faster but
approximate.
#install.packages("Rtsne")
library(Rtsne)
## plot samples (not interesting)
TS1 = Rtsne(t(mRNA$X),perplexity=3,max_iter=2000)$Y
plot(TS1[,1],TS1[,2],col=as.integer(mRNA$meta$time),pch=19,cex=2, main="tSNE for samples (reducing gene space)")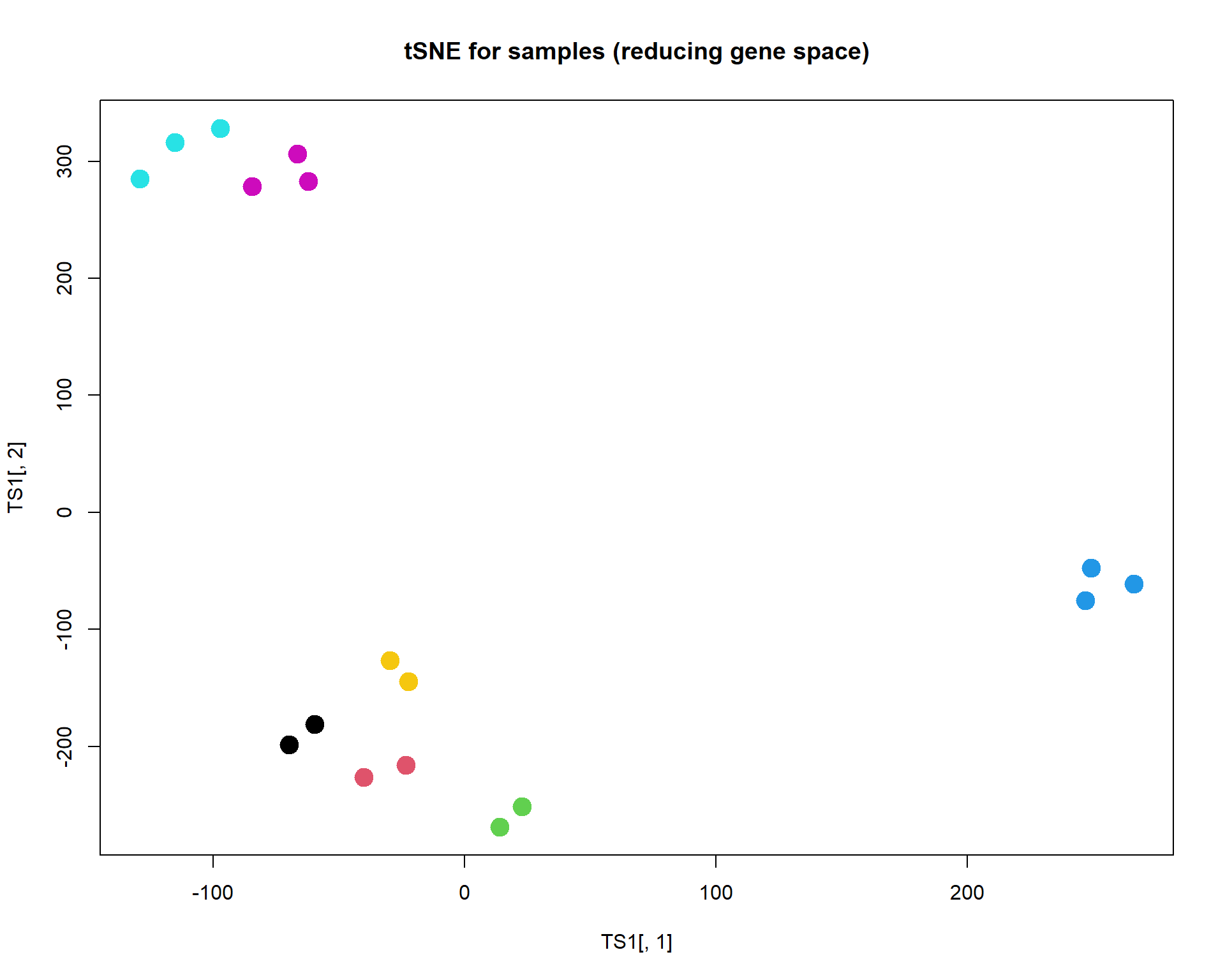
## plot features
TS2x = Rtsne(mRNA$X,perplexity=20,max_iter=1000,check_duplicates = FALSE )$Y
plot(TS2x[,1],TS2x[,2],col="#00AA0033",pch=19,cex=size, main="tSNE for genes (original)")
text(TS2x[genes,1],TS2x[genes,2],names(genes),col=1,pch=19,font=2)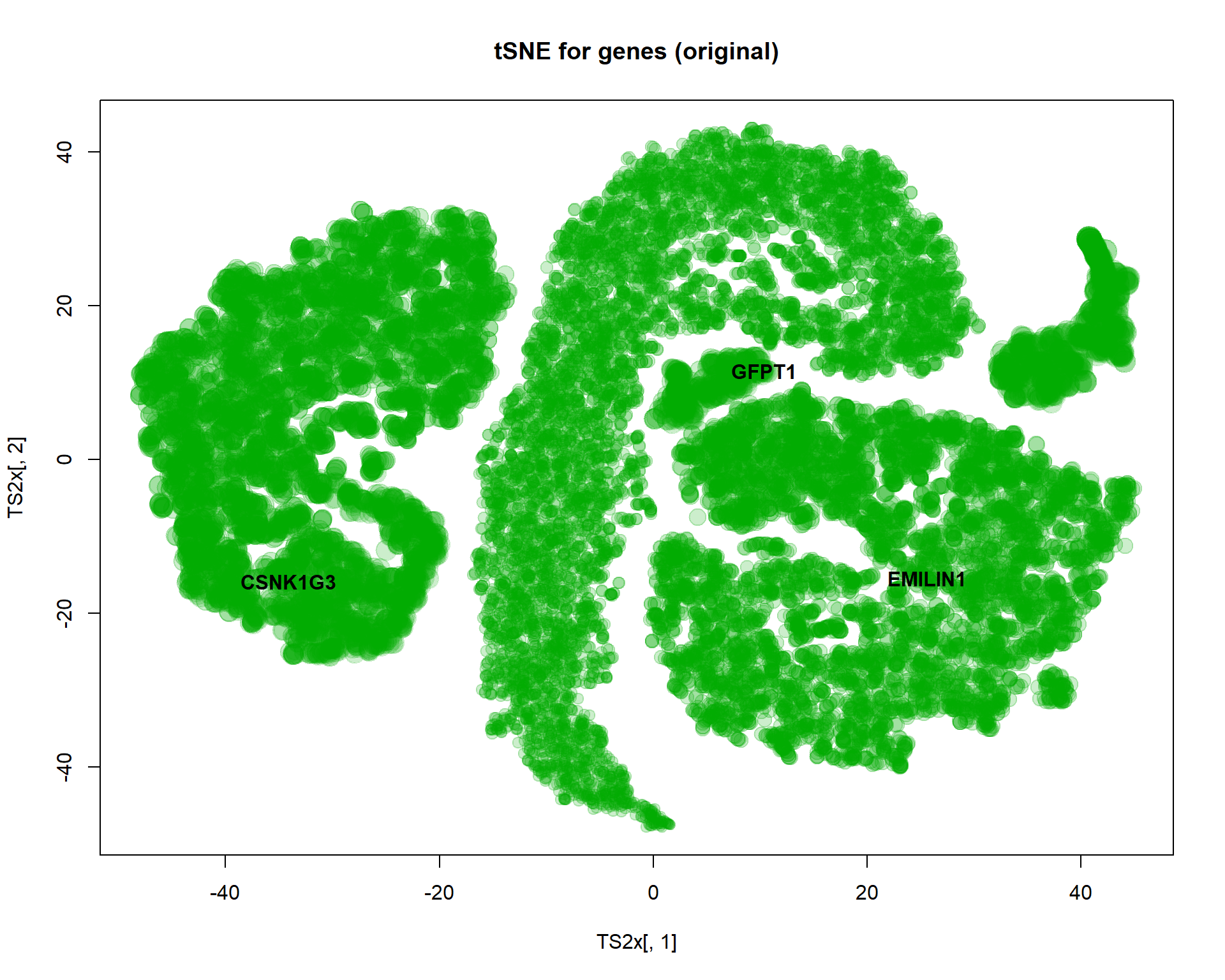
## same for scaled features (Z was calculated earlier)
TS2z = Rtsne(Z,perplexity=50,max_iter=1000,check_duplicates = FALSE)$Y
plot(TS2z[,1],TS2z[,2],col="#00AA0033",pch=19,cex=size, main="tSNE for genes (standardized genes)")
text(TS2z[genes,1],TS2z[genes,2],names(genes),col=1,pch=19,font=2)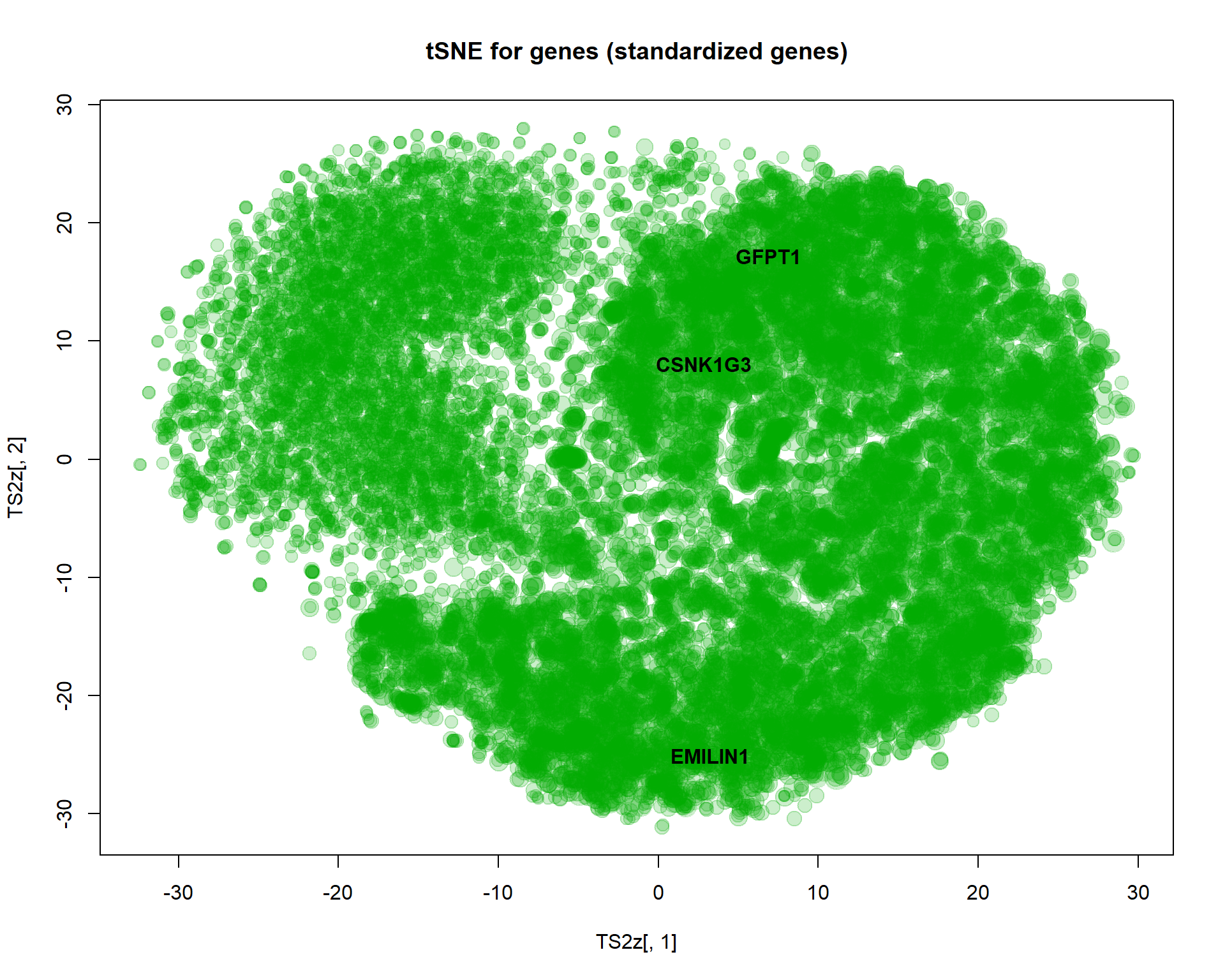
There is another package - tsne. It is
slower, but in some cases more precise. If “precise” can be used for
describing t-SNE ;)
You can also use tSNE and UMAP from plotPCA:
source("http://r.modas.lu/plotPCA.r")
par(mfcol=c(1,2))
plotTSNE(mRNA$X,col=as.integer(mRNA$meta$time),perplexity=5,cex.names=1,cex=2,main="tSNE")
plotUMAP(mRNA$X,col=as.integer(mRNA$meta$time),cex.names=1,cex=2,main="UMAP")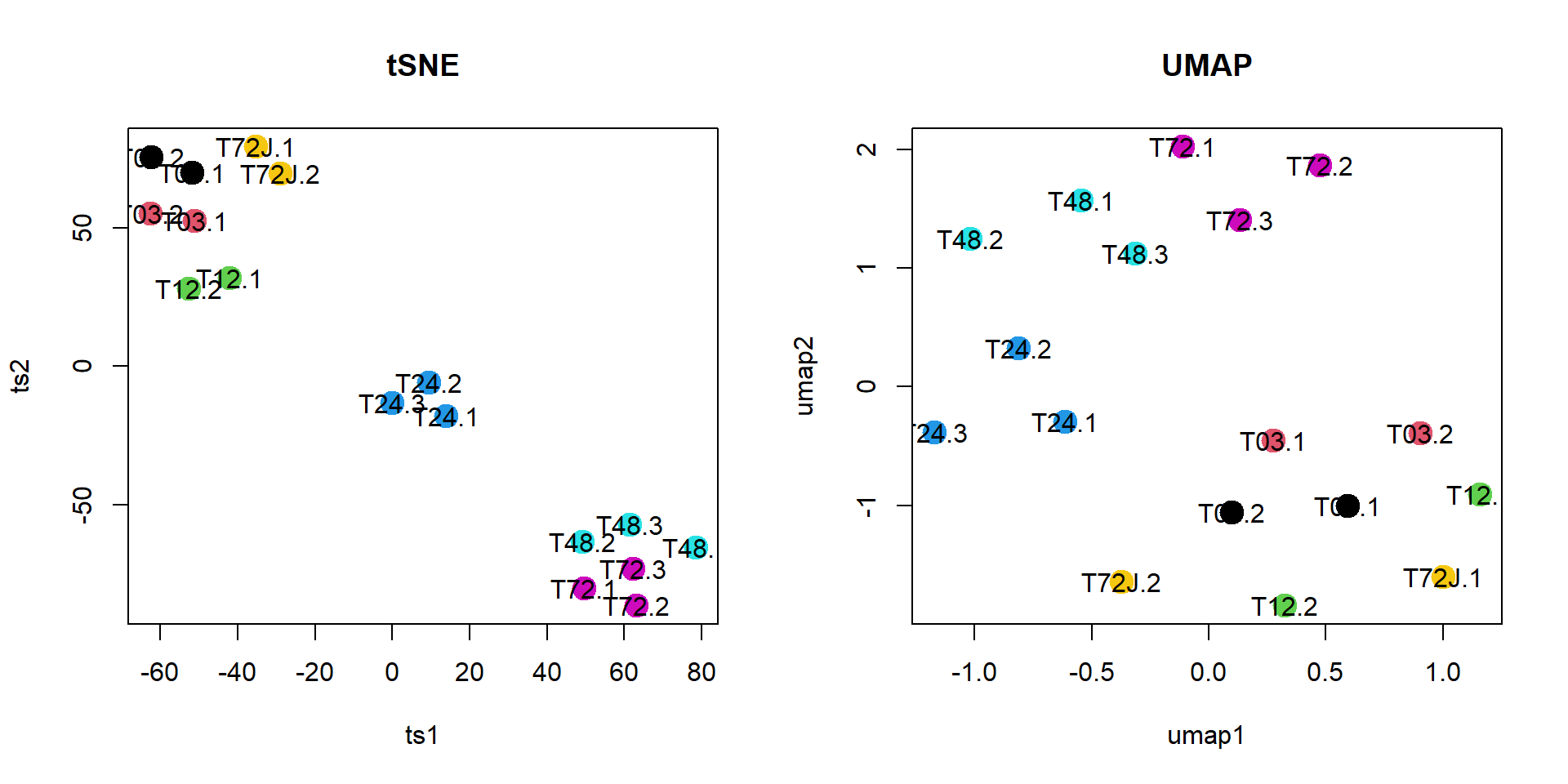
Exercises
- Build PCA for miR data. Access to the data -
miR$X. Sample annotation -miR$meta
prcomp()andt(),plotPCA()
- Build PCA for mice from Mice data and identify ourliers. Use the data frame without 3 first columns:
Mice[,-(1:3)]
prcomp()andtext(); orplotPCA()andt()
- Build MDS for miR data.
dist(),t(),cmdscale()
- Build tSNE for
irisdata. Use all columns except 5th for calculation, and 5th - for coloring.
Rtsne()
| Prev Home Next |
By Petr Nazarov
|