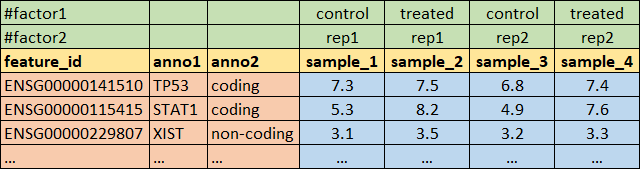
1.1. Main data formats
In bioinformatics people prefer using plain text format for the data (obvious reason…). Main formats are TSV and CSV.
txt, tsv - tab-separated values:
read.table()orread.delim()csv - comma-separated values:
read.csv()orread.table(...,sep=",")orread.delim()json - JavaScript Object Notation - format used for storing and transporting data in text format:
rjson::fromJSON(not very often)
Some binary formats you may easily use as well.
xls, xlsx - Excel 2003 or 2010:
readxl::read_excel()RData, Rda - R environment (variables and their names):
load()RDS - stores a single R object (without its name):
loadRDS()
1.1.1. Text tables: TXT, TSV, CSV
Example TSV:
## let's read this file from internet
Mice = read.table("http://edu.modas.lu/data/txt/mice.txt",
sep = "\t",
header= TRUE,
stringsAsFactors = TRUE)
str(Mice)## 'data.frame': 790 obs. of 14 variables:
## $ ID : int 1 2 3 368 369 370 371 372 4 5 ...
## $ Strain : Factor w/ 40 levels "129S1/SvImJ",..: 1 1 1 1 1 1 1 1 1 1 ...
## $ Sex : Factor w/ 2 levels "f","m": 1 1 1 1 1 1 1 1 1 1 ...
## $ Starting.age : int 66 66 66 72 72 72 72 72 66 66 ...
## $ Ending.age : int 116 116 108 114 115 116 119 122 109 112 ...
## $ Starting.weight : num 19.3 19.1 17.9 18.3 20.2 18.8 19.4 18.3 17.2 19.7 ...
## $ Ending.weight : num 20.5 20.8 19.8 21 21.9 22.1 21.3 20.1 18.9 21.3 ...
## $ Weight.change : num 1.06 1.09 1.11 1.15 1.08 ...
## $ Bleeding.time : int 64 78 90 65 55 NA 49 73 41 129 ...
## $ Ionized.Ca.in.blood : num 1.2 1.15 1.16 1.26 1.23 1.21 1.24 1.17 1.25 1.14 ...
## $ Blood.pH : num 7.24 7.27 7.26 7.22 7.3 7.28 7.24 7.19 7.29 7.22 ...
## $ Bone.mineral.density: num 0.0605 0.0553 0.0546 0.0599 0.0623 0.0626 0.0632 0.0592 0.0513 0.0501 ...
## $ Lean.tissues.weight : num 14.5 13.9 13.8 15.4 15.6 16.4 16.6 16 14 16.3 ...
## $ Fat.weight : num 4.4 4.4 2.9 4.2 4.3 4.3 5.4 4.1 3.2 5.2 ...Example CSV:
BC = read.csv("http://edu.modas.lu/data/txt/breastcancer.csv",
comment.char="#",
header= TRUE)
str(BC)## 'data.frame': 699 obs. of 11 variables:
## $ sample : int 1000025 1002945 1015425 1016277 1017023 1017122 1018099 1018561 1033078 1033078 ...
## $ clump.thickness: int 5 5 3 6 4 8 1 2 2 4 ...
## $ uni.size : int 1 4 1 8 1 10 1 1 1 2 ...
## $ uni.shape : int 1 4 1 8 1 10 1 2 1 1 ...
## $ adhesion : int 1 5 1 1 3 8 1 1 1 1 ...
## $ epith.size : int 2 7 2 3 2 7 2 2 2 2 ...
## $ bare.nuclei : int 1 10 2 4 1 10 10 1 1 1 ...
## $ bland.chromatin: int 3 3 3 3 3 9 3 3 1 2 ...
## $ normal.nucleoli: int 1 2 1 7 1 7 1 1 1 1 ...
## $ mitoses : int 1 1 1 1 1 1 1 1 5 1 ...
## $ class : int 0 0 0 0 0 1 0 0 0 0 ...1.1.2. Excel
Example Excel:
# install.packages("readxl")
library(readxl)
## download Excel file and store it locally
download.file("http://edu.modas.lu/data/xls/cancer.xlsx",destfile="cancer.xlsx",mode = "wb")
## read Excel file into `tibble`
Cancer = read_excel("cancer.xlsx")
str(Cancer)
## optional: change to `data.frame`
Cancer = as.data.frame(Cancer)1.1.3. JSON
JSON (JavaScript Object Notation) is an open standard file format and data interchange format that uses human-readable text to store and transmit data objects consisting of attribute–value pairs, arrays, etc. It is a commonly used data format with diverse uses in electronic data interchange, including that of web applications with servers [Wikipedia].
## load the package "rjson"
#install.packages("rjson")
library("rjson")
## read the file
coad = fromJSON(file="http://edu.modas.lu/data/txt/clin_coad.json")
## check number of records
length(coad)## [1] 2937## check record #1
str(coad[[1]])## List of 6
## $ exposures :List of 1
## ..$ :List of 5
## .. ..$ alcohol_history : chr "Not Reported"
## .. ..$ updated_datetime: chr "2019-07-31T17:59:51.385908-05:00"
## .. ..$ exposure_id : chr "5fff4884-4d50-58c2-98eb-29a8fd008e50"
## .. ..$ submitter_id : chr "TCGA-DC-6158_exposure"
## .. ..$ state : chr "released"
## $ case_id : chr "0011a67b-1ba9-4a32-a6b8-7850759a38cf"
## $ project :List of 1
## ..$ project_id: chr "TCGA-READ"
## $ submitter_id: chr "TCGA-DC-6158"
## $ diagnoses :List of 1
## ..$ :List of 26
## .. ..$ synchronous_malignancy : chr "No"
## .. ..$ ajcc_pathologic_stage : chr "Stage I"
## .. ..$ days_to_diagnosis : num 0
## .. ..$ treatments :List of 2
## .. .. ..$ :List of 7
## .. .. .. ..$ updated_datetime : chr "2019-07-31T17:59:51.385908-05:00"
## .. .. .. ..$ submitter_id : chr "TCGA-DC-6158_treatment_1"
## .. .. .. ..$ treatment_id : chr "c3bee975-4617-54ab-aca2-966e0465dc00"
## .. .. .. ..$ treatment_type : chr "Pharmaceutical Therapy, NOS"
## .. .. .. ..$ state : chr "released"
## .. .. .. ..$ treatment_or_therapy: chr "no"
## .. .. .. ..$ created_datetime : chr "2019-04-28T10:50:51.230930-05:00"
## .. .. ..$ :List of 6
## .. .. .. ..$ updated_datetime : chr "2019-07-31T17:59:51.385908-05:00"
## .. .. .. ..$ submitter_id : chr "TCGA-DC-6158_treatment"
## .. .. .. ..$ treatment_id : chr "c89f33fd-056e-5333-bad0-4ea7b6053517"
## .. .. .. ..$ treatment_type : chr "Radiation Therapy, NOS"
## .. .. .. ..$ state : chr "released"
## .. .. .. ..$ treatment_or_therapy: chr "no"
## .. ..$ last_known_disease_status : chr "not reported"
## .. ..$ tissue_or_organ_of_origin : chr "Rectum, NOS"
## .. ..$ days_to_last_follow_up : num 216
## .. ..$ age_at_diagnosis : num 25842
## .. ..$ primary_diagnosis : chr "Adenocarcinoma, NOS"
## .. ..$ updated_datetime : chr "2023-10-06T12:21:52.117337-05:00"
## .. ..$ prior_malignancy : chr "no"
## .. ..$ year_of_diagnosis : num 2010
## .. ..$ prior_treatment : chr "No"
## .. ..$ state : chr "released"
## .. ..$ ajcc_staging_system_edition: chr "7th"
## .. ..$ ajcc_pathologic_t : chr "T2"
## .. ..$ morphology : chr "8140/3"
## .. ..$ ajcc_pathologic_n : chr "N0"
## .. ..$ ajcc_pathologic_m : chr "M0"
## .. ..$ submitter_id : chr "TCGA-DC-6158_diagnosis"
## .. ..$ classification_of_tumor : chr "not reported"
## .. ..$ diagnosis_id : chr "42b633c0-1e17-52c1-82c7-82c677d5b230"
## .. ..$ icd_10_code : chr "C20"
## .. ..$ site_of_resection_or_biopsy: chr "Rectum, NOS"
## .. ..$ tumor_grade : chr "Not Reported"
## .. ..$ progression_or_recurrence : chr "not reported"
## $ demographic :List of 12
## ..$ demographic_id : chr "82a0689e-a9b0-55dc-96c2-f463bed2d317"
## ..$ ethnicity : chr "not hispanic or latino"
## ..$ gender : chr "male"
## ..$ race : chr "white"
## ..$ vital_status : chr "Dead"
## ..$ updated_datetime: chr "2019-07-31T17:59:51.385908-05:00"
## ..$ age_at_index : num 70
## ..$ submitter_id : chr "TCGA-DC-6158_demographic"
## ..$ days_to_death : num 334
## ..$ days_to_birth : num -25842
## ..$ state : chr "released"
## ..$ year_of_birth : num 1940